신약개발과정에서의 
약물대사 연구
한국생명공학연구원 오수진 선임연구원
1. 서론
신약개발은 많은 시간과 노력 그리고 높은 비용을 소모하는 과정이지만 높은 부가가치를 창출할 수 있는 산업이다. 신약개발을 위한 임상연구 과정에서의 주요 실패요인이 약물대사 및 약물동태 (pharmacokinetics, PK)로 알려진 1990년대 초반 이후 신약개발의 초기단계에서부터 ADME (Absorption, Distribution, Metabolism, Excretion) 평가연구를 적극적으로 도입함으로써 신약개발 성공률을 높이기 위한 노력이 지속적으로 진행되어 왔고 현재 다국적제약사를 중심으로 신약개발의 일반적인 흐름으로 자리 잡고 있다. 이러한 노력의 결과로 ADME의 문제로 인한 신약개발 실패는 현저히 감소되었다. 특히, 신약개발과정에서 후보물질의 대사적 특성을 초기에 연구하는 것은 신약개발의 비용과 시간을 절약하는 주요한 전략으로 자리 잡았다.
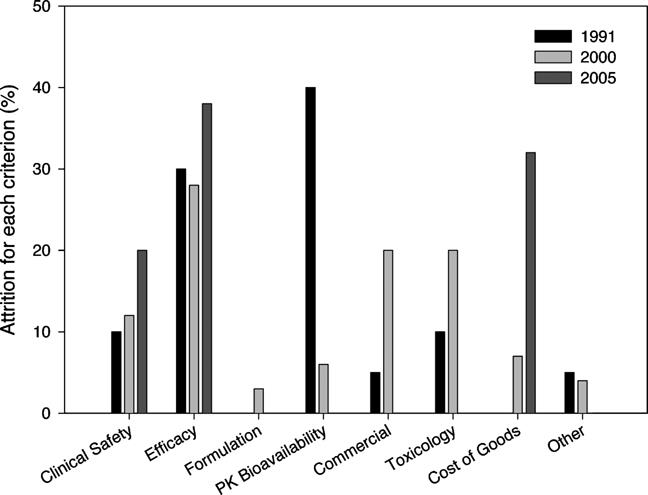
<신약개발과정에서의 실패요인 분석> (Ref. 1)
현재 신약개발 과정에서는 초기단계인 신약탐색단계(drug discovery)에서부터 활성(activity)뿐만 아니라 물성(property)에 대한 최적화 연구도 동시에 진행함으로써 임상에서의 성공가능성이 높은 ‘drug-like property’를 지닌 후보물질 도출을 위하여 노력하고 있다. Drug-like한 특성은 선도물질(lead compound) 도출 이후 선도물질의 최적화 과정에서 그 중요성이 더욱 강조되며 그 이유는 후보물질(candidate)의 임상에서의 성공 가능성을 예측하는 지표가 될 수 있기 때문이다. Lipinski는 drug-like한 특성에 대해 임상1상까지 개발중인 물질이 살아남을 만큼 충분한 ADME/Tox 의 성질을 가지는 것이라 정의하였으며 현재 구조적, 물리화학적, ADME/Tox 측면에서 drug-likeness 와 관련된 평가를 신약개발의 초기단계에 수행하고 있다 (Ref 2).
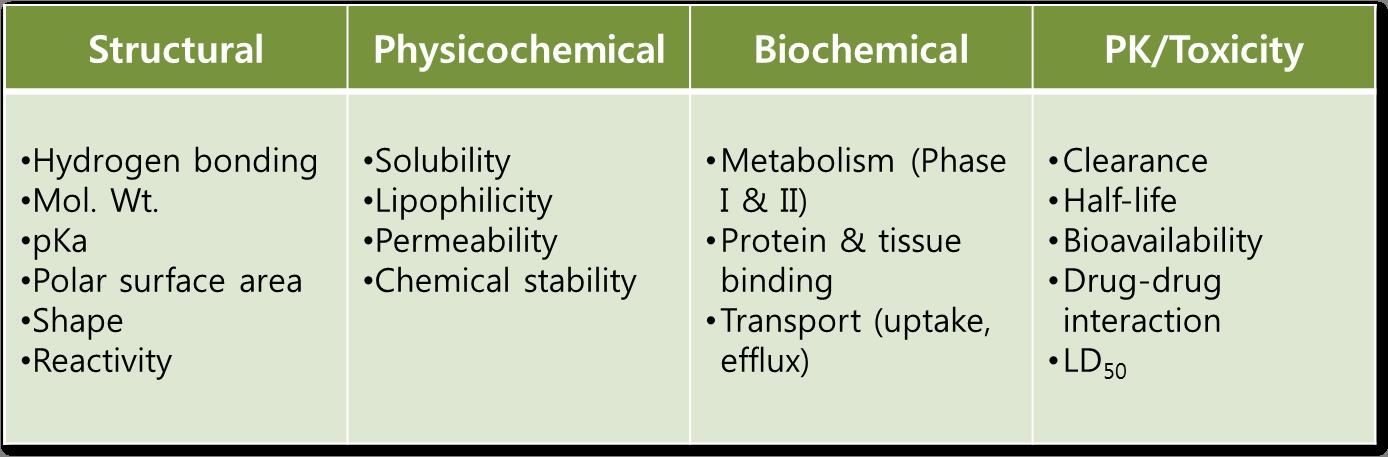 (Ref. 2)
(Ref. 2)
2. 선도물질의 최적화와 후보물질 도출을 위한 약물대사연구
신약개발 초기단계에서 도출된 선도물질들은 일반적으로 약으로 갖춰야 할 적절한 물성을 지니지 못한다. 시험관에서 아무리 좋은 효과를 나타낸 물질이라도 동물에 투여 후 충분한 혈중 농도를 지속적으로 유지하지 못한다면 신약으로의 가치를 잃게 된다. 신약개발 초기단계에서 DMPK(drug metabolism and pharmacokinetics)를 연구하는 사람들이 당면하는 주요 문제는 낮은 흡수율, 낮은 혈중 농도, 신속한 대사 및 배설 등과 같은 바람직하지 못한 PK 특성들로 많은 DMPK 연구자들은 물질의 PK 특성을 개선하여 선도물질을 최적화하고 후보물질을 도출하기 위해 다양한 노력을 기울인다. 특히, 신약개발 초기단계에서의 약물대사연구는 개발중인 약물의 운명을 결정하는데 매우 중요한 역할을 담당한다. 약물이 효과를 나타내기 위해서는 그 물질이 표적(target)에 무사히 도달하여 약효를 내기 충분한 농도에 도달하여야 하는데 약물이 대사되면 특별한 경우를 제외하고는 대부분 활성을 잃기 때문에 약을 개발하는 입장에서 약물대사는 넘어야 할 산인 것이다.
(1) 약물대사(Drug metabolism, Biotransformation) 및 대사안정성 시험
약물 대사는 거의 모든 장기(예. 장, 폐 및 신장)에서 일어날 수 있으나 주요 약물대사효소는 간에 높은 수준으로 발현되어 있어 간이 약물대사에 있어 가장 중요한 장기라 할 수 있다. 대사과정은 흡수된 외인성 물질의 수용성을 증가시켜 체외배설이 용이한 형태로 전환시키는 과정으로 약을 포함한 외인성 물질의 체내 축적을 막는 중요한 방어체계라 할 수 있다. 하지만 때로는 약물대사의 과정이 불완전하여 대사과정에서 오히려 독성이 증가하는 bioactivation이 유발되기도 한다.
일반적으로 약물대사반응은 Phase I 과 Phase II 반응으로 나뉜다. Phase I 반응은 전형적으로 관능기(functional group, 예. hydroxyl)를 도입하여 물질의 극성(polarity)를 증가시키는 반응이고 Phase II 반응은 모화합물 자체 또는 모화합물이 Phase I 반응을 통해 polar한 관능기가 도입된 후 생성된 대사체에 일어나는 conjugation 반응으로 수용성을 증가시킨다. Phase I 반응은 주로 cytochrome P450(CYP) enzymes에 의한 산화반응(oxidation)이 주된 반응이며 가수분해 (hydrolysis)와 환원반응 (reduction)도 가능하다. Phase II 반응은 glucuronidation, sulfation, glutathione conjugation, amino acid conjugation, methylation 및 acetylation 반응이 있으며 methylation과 acetylation을 제외한 다른 반응은 모두 수용성을 증가시키는 반응이다.
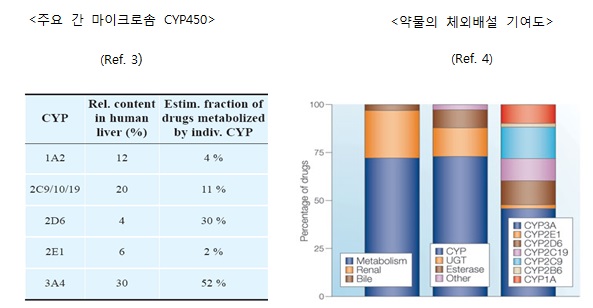
생체 내로 흡수된 약물이 몸 밖으로 배설되는 경로의 총합을 약물 청소율 (drug clearance)이라 한다. 약물 청소율은 주로 뇨 또는 변으로의 배설과 약물대사에 의해 일어나는데 시판되고 있는 약물 중 약 75% 정도가 약물대사경로를 통해 몸 밖으로 제거된다고 알려져 있다. 즉, 약물대사가 약물 청소율에 있어 매우 중요한 경로임을 알 수 있다. 또한 약물대사의 75% 정도는 CYP에 의해 일어나는 것으로 보고되었는데 이는 시판되는 약물의 약 55%가 CYP 에 의해 매개된다는 것을 의미한다. CYP는 매우 다양한 동종효소(isoform)를 가지는데 특히 5가지 동종효소, CYP 1A2, CYP 2C9, CYP 2C19, CYP 2D6 및 CYP 3A4가 전체 CYP에 의한 대사의 90% 이상을 차지한다고 보고되었다. 이러한 이유 때문에 신약개발 초기 스크리닝 단계에서 약물대사에 안정한 특히 CYP에 보다 안정한 물질을 찾고자 하는 노력의 일환으로 microsomes을 이용한 대사 안정성 시험을 수행한다. 앞서 언급한 것처럼 약물대사는 주로 간에서 일어나기 때문에 약물대사와 관련한 in vitro 시험은 간으로부터 유래한 tissue fraction (예. microsomes, cytosol 또는, S9 fraction) 및 primary hepatocyte 등을 enzyme source로 사용하는 것이 일반적이다.
(2) 대사체 동정(Metabolite identification)
현재 신약개발과정에서는 신약개발의 각 단계를 효율적으로 지원할 수 있는 다양한 in vitro 및 in vivo 시험계가 개발되어 활용되고 있다. 특히 선도물질의 최적화 단계에서 약물대사연구는 더욱 큰 의미를 지닌다. 특히, 약물 대사체의 동정 및 대사경로의 규명은 신약개발과정에서 필수적으로 요구되며 신약개발 규제/허가기관에서도 물질의 안전성 및 유효성 검증을 위하여 필수적으로 요구하고 있다(Ref. 5).
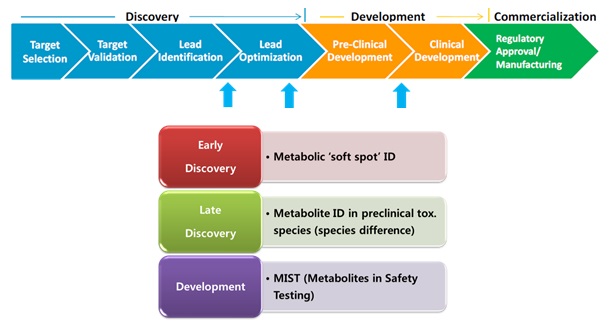
약물 대사체의 동정은 신약개발단계에 따라 그 목적을 달리하는데 신약개발 초기단계에서는 주로 microsomes과 hepatocyte 등 in vitro 시험계를 이용하여 수행되며 개발중인 물질의 구조 중 대사에 취약한 부분(metabolic soft spot)을 찾아 화학자들에게 정보를 제공하여 보다 대사에 안정한 물질을 합성할 수 있도록 하는 것을 그 목적으로 한다. 후보물질의 평가단계에서는 다양한 실험동물 종(species)에서의 약물 대사체 스크리닝을 통해 인체와 유사한 대사체 프로파일을 보이는 종을 찾고 그 실험동물 종을 이용하여 전임상 시험을 수행할 수 있게 함으로써 전임상 실험결과와 임상실험 결과간의 상관관계를 높이는데 주안점을 두고 있다. 2008년도 미국 FDA(Food and Drug Administration)에서 MIST (Metabolites in Safety Testing)라는 guidance를 발표하였다. 이 guidance에서는 대사체의 AUC (area under the curve)가 모화합물(parent compound) AUC의 10% 이상이고 그 대사체가 실험에 사용한 동물 종에서 발견되지 않거나 인간에 비해 낮은 농도로 측정될 경우 모약물 외에 대사체의 전임상 독성을 요구하고 있다. 또한 인간에 선택적인 대사체가 일정량 이상 생성될 경우 신약개발을 위해서는 대사체의 안전성을 필수적으로 평가해야 됨을 강조하고 있다. 이러한 이유로 많은 제약회사에서는 임상시험에서 발생할 수 있는 예상치 못한 독성의 가능성을 최소화하기 위해 후보물질 평가 및 개발단계에서 대사체의 종차를 정성 및 정량적 측면 모두에서 평가한다. 신약개발 초기단계에서의 대사체 연구가 PK 특성을 개선하기 위한 부분에 목적을 두어 정성적 분석에 초점을 맞추었다면 후기단계에서는 독성적 측면에서의 대사 연구를 수행하기 위해 정성뿐 아니라 정량적 분석이 강조되고 있다.
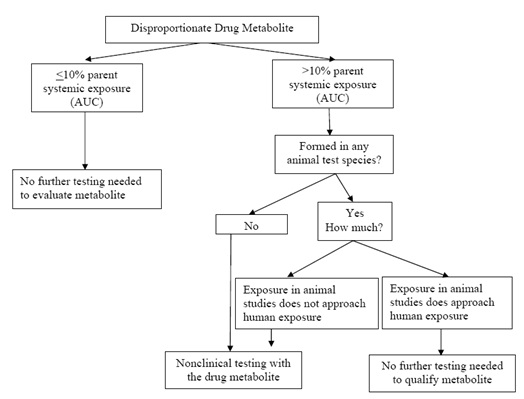
<대사체 안전성시험을 위한 decision tree> (Ref. 5)
(2) 약물유래 간독성과 약물상호작용
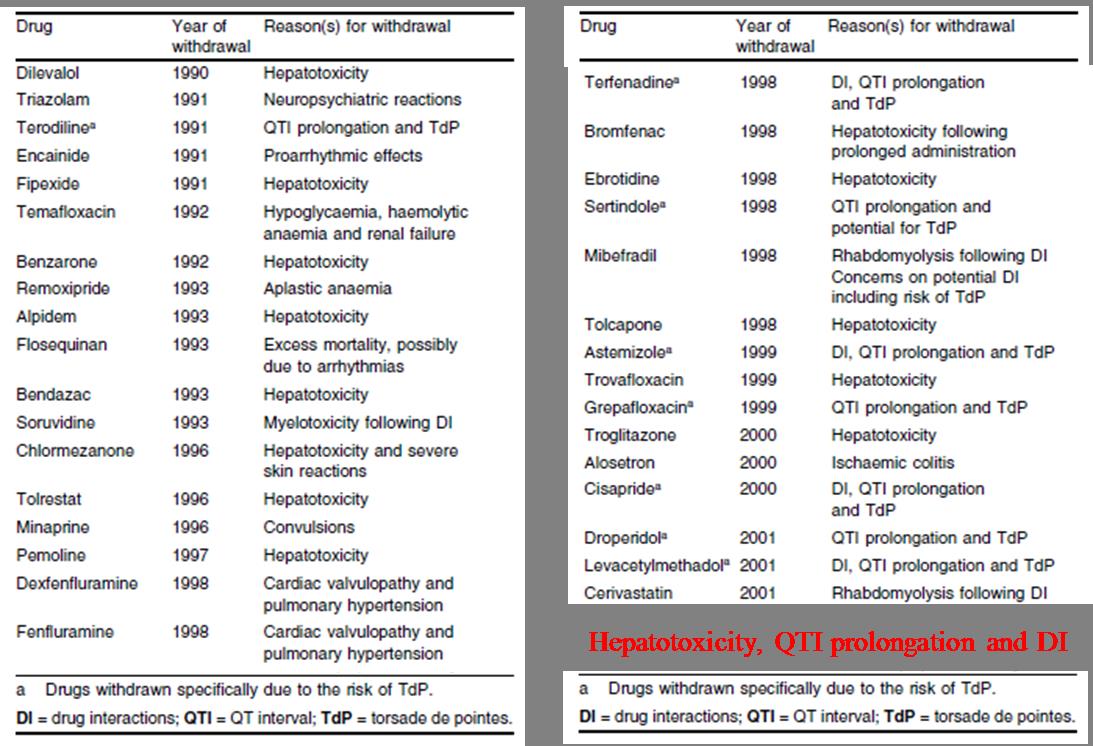
1990년 이후 독성문제로 시판 중에 시장에서 철수한 33종의 의약품을 원인 별로 분석해본 결과 13종(약40%)은 간독성, 10건(약30%)은 QT interval 증가에 따른 심장독성(부정맥) 그리고 약물대사를 통한 약물상호작용 8건(약24%)으로 조사되었다(Ref. 6). 심장독성을 제외하고 간독성과 약물상호작용은 약물대사와 매우 밀접한 관련을 지닌다. 약물유래 간독성(DILI; drug-induced liver injury)은 모약물 보다는 친전자성의 독성 대사체에 기인한다. 1990년대 이후 benoxaprofen, iproniazid, nefazodone, tienilic acid, troglitazone, bromfenac (unclear) 등의 약물은 대사체에 의한 독성으로 시장에서 철수하였으며, black box 경고가 부가된 15개의 약물 중 8개(dacarbazine, dantrolene, felbamate, flutamide, isoniazid, ketoconazole, tolcapone, valproic acid) 역시 독성 대사체에 기인하는 것으로 알려졌다. 심장독성의 경우 심장의 action potential을 조절하는 ion channel, 특히 hERG와 같은 중요한 표적 단백질이 규명되어 신약개발의 초기단계에서 심장독성에 대한 평가가 진행되고 있으나 약물유래 간독성은 표적 단백질이 발굴되지 않고 있어 신약개발의 초기단계에서 간독성을 평가할 수 있는 연구방법은 매우 제한적이다. 결과적으로 약물유래 간독성은 현재까지도 신약개발의 가장 큰 허들 중 하나이다.
<1990년 이후 의약품 시장 철수 요인> (Ref. 6)
약물상호작용(drug interaction)에 의해 시장에서 퇴출된 대표적인 약물은 Terfenadine으로 1990년 심각한 약물상호작용에 대한 보고가 있었고 1992년 미국 FDA 에서 black box 경고가 부가된 이후 1998년 시장에서 퇴출되었다. 의약품의 안전성 평가에서 약물상호작용은 한가지 약물이 다른 약물(또는 내⋅외인성 물질과 생체 구성성분)들과 상호작용하여 투여된 약물의 약효나 독성에 변화가 유발되는 현상을 말한다. 약물대사를 통한 약물상호작용은 A 약물이 B 등 다른 약물의 체내 동태(혈중 또는 조직에서 AUC, 반감기, 최대농도 등)가 변동되고 결과적으로 B 약물의 약효/독성이 정상과 다르게 발현하는 것이다. 앞서 약물대사에 있어 CYP의 중요성에 대해 언급하였는데 CYP의 큰 특징은 외인성 물질의 노출에 의해 발현이나 활성이 유도되거나 억제되는 특징을 가지고 있다. CYP의 유도는 약물의 농도를 감소시켜 약효가 나타날 수 있는 유효농도에 도달하지 못하게 하는 반면 CYP의 억제는 약물의 농도를 증가시켜 약효를 넘어 독성을 유발하게 할 수 있다. 약물대사를 통한 의약품의 상호작용은 의약품과 의약품 외에도 식품과 의약품, 천연물과 의약품 등 다양한 조합에서도 발생할 수 있다. 약물상호작용에서 CYP를 유도하거나 억제하여 병용 처리된 다른 의약품의 PK 지표를 변화시키는 것을 가해자(perpetrator) 그리고 변화된 CYP에 의해 대사를 받는 것을 피해자(victim or substrate)로 표현할 수 있다. 약물상호작용으로 혈중 농도가 증가하여 독성을 유발하는 의약품은 주로 CYP의 기질인 피해자이다.
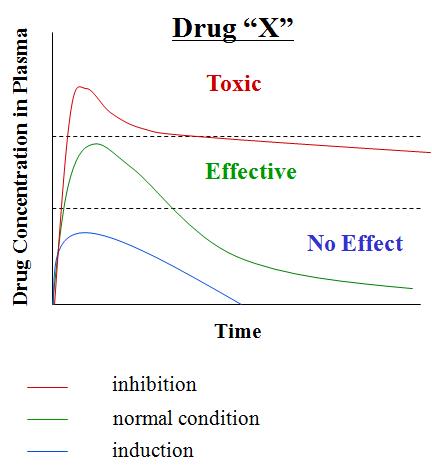
<약물상호작용의 결과>
현재 신약개발 과정 중 인체 내 약물상호작용을 예측하고 그 기전을 규명하기 위해 CYP inhibition/induction 시험을 routine하게 수행하고 있다. 최근 2012년에 미국 FDA에서 업데이트된 가이드라인을 발표하였다. 그 주요 내용을 살펴보면 in vitro CYP inhibition 연구의 경우, 기존의 5종의 CYPs, 즉, CYP1A2, CYP2C9, CYP2C19, CYP2D6 및 CYP3A4 에 대한 CYP inhibition study에서 CYP2B6, CYP2C8 2종을 추가하여 7종의 CYP 에 대한 inhibition study를 수행할 것을 권장하고 있으며 inhibition의 mechanism을 이해하기 위해 좀더 자세한 reversible inhibition/time dependent inhibition(TDI) 에 대한 기준을 제시하고 있다. in vitro CYP induction의 경우 새로운 end point로 mRNA의 변화를 제시하였고 in vivo study를 대신할 PBPK modeling 및 simulation과 관련된 내용을 포함하고 있다. 또한 주요 배설경로(elimination pathway)에 대한 연구에서 기존에 CYP 에 대한 reaction phenotyping 연구에 새롭게 UGT(UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7 및 UGT2B15)에 대한 평가를 추가하였다. 그리고 기존의 약물대사효소, 특히 CYP에 대한 약물상호작용 연구에서 transport의 약물상호작용에 대한 내용을 추가한 것이 가장 큰 변화라 할 수 있다(Ref. 7). 종합적으로 미국 FDA에서 2012년에 발표된 guidance는 약물상호작용에 대한 광범위한 결과를 요구하고 있다.
3. 결론
신약개발 과정에서 수행되고 있는 약물대사의 연구는 개발중인 물질의 PK 특성과 독성적 측면을 이해하기 위함이다. 신약개발의 궁극적인 목적은 사람에서 치료제로 사용할 수 있는 물질의 개발이라 볼 수 있다. 이를 위해서는 반드시 규제기관으로부터 허가를 받아야 한다. 규제기관의 허가를 받을 때 가장 크게 고려되는 점은 약효와 안전성의 문제이며 이에 대한 이해를 돕기 위해 DMPK 연구가 수행 되어야 하는 것이다. 실제로 DMPK 관련 정보를 이용하여 약물의 화학구조, 효능 및 PK 가 적절하게 균형을 이룰 수 있는 약물 설계를 통해 성공한 사례는 다수 존재한다. 비록 임상시험에 앞서 in vitro 및 실험동물을 이용한 다양한 DMPK 시험을 수행하고 있고 이러한 결과가 매우 유용한 정보를 제공하지만 human prediction에 있어서는 분명 한계가 있다. 따라서 선행연구로부터 얻은 결과를 사람에 외삽(extrapolation) 할 때에는 많은 주의가 필요하다. 실험동물 간 생리적인 특성이 유사하다 하더라도 특히 약물대사의 경우 약물대사효소의 종차(species difference)가 매우 크기 때문에 실험동물 결과로부터 사람에서 신뢰할 만한 결과를 예측 하기 위해서는 무엇보다도 종간 유사점과 차이점에 관련된 mechanism의 이해가 중요하다 할 수 있다. 신약개발 과정은 다양한 분야의 전문가가 모여 신규 치료제의 개발이라는 하나의 목표를 향해 나아가는 긴 여정이다. 아프리카 속담에 ‘빨리 가려면 혼자 가고 멀리 가려면 함께 가라’라는 말이 있다. 신약개발이라는 긴 여정의 끝에 성공이라는 열매를 얻기 위해 노력하는 많은 이들이 크게 공감할 수 있는 말이라 생각한다.
References
1. Ruiz-Garcia A. Bermejo M. Moss A. and Casabo VG. (2008) Pharmacokinetics in drug discovery. J. Pharm. Sci. 97:654-90.
2. Kerns EH, Di Li. (2008) Drug-like properties: Concepts, Structure Design and Methods: from ADME to Toxicity Optimization.
3. Zuber R, Anzenbacherova E, Anzenbacher P. 2002. Cytochromes P450 and experimental models of drug metabolism. J Cell Mol 6(2): 189-98.
4. Wienkers LC, Heath TG. (2005) Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov 4(10):825-33.
5. U.S. FDA (2008) Guidance for industry. Safety testing for drug metabolites.
6. Shah RR. (2004) Pharmacogenetic aspects of drug-induced torsade de pointes: potential tool for improving clinical drug development and prescribing. Drug Saf. 27:145-72.
7. U.S. FDA. (2012) Guidance for industry. Drug Interaction studies-Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations.

