성공적인 글로벌 의약품 개발 및 허가. 등록을 위해 참고할만한 요소
(주)유니테스 대표 조제영
신약개발은 성공 시 높은 수익을 창출하지만 막대한 투자비와 많은 시간이 소요되며 성공률이 매우 낮은 고 위험 사업이다. 그러나 국내 제약산업의 매출규모와 R&D 투자비 그리고 기술력은 아직 영세한 수준이고, 세계적인 선도기업 및 블록버스터 의약품 창출에 대한 경쟁력은 취약한 상황으로 글로벌신약의 개발을 목표로 하는 과정에 참고하면 도움이 될만한 요소에 대하여 소개하고자 한다.
1. 초기 기획의 단계부터 진행단계 그리고 마지막 허가단계까지 Target Product Profile (TPP) 을 염두에 둔 개발을 시행하여야 한다.
TPP 는 약물의 Labeling 이다. 실질적으로 약물의 전 개발과정은 약물의 판매를 위한 허가를 받을 때 Labeling 을 작성하기 위한 Supporting 자료를 만드는 과정이다. 달리 표현하여 TPP 는 기말고사를 준비하는 학생들의 시험문제지에 해당한다. 허가등록까지의 긴 개발과정속에 수행하여야 하는 연구들은 구성 항목의 맹목적 시행이 아니라 TPP 라는 목표를 바탕으로 TPP 구성을 위하여 목적지향적인 시험의 디자인, 진행 및 자료의 규합이 이루어지도록 진행되어야 한다.
최종 목표인 Goal (TPP) 를 바탕으로 끊임 없는 단계별 Back and Forth 연계 작업이 이루어져야 한다.
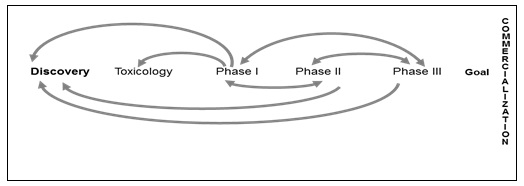
TPP 를 구성하는 요소 중 약물이 치료하고자 하는 적응증을 Label 에 넣고자 한다면 비임상 및 임상시험에서 치료하고자 하는 적응증에 대한 약물의 반응성이 있음이 증빙되어야 하며, Contraindication 에 대한 정보를 Label 에 넣어 마켓에서 안전한 약물의 처방 및 복용이 이루어지고자 하면 비임상 및 임상의 과정 중에 발견되는 독성현상 및 부작용이 면밀히 검토되고 정리되어 이를 바탕으로 왜 contrainidication 의 항목이 Label 에 반영되어 판매 후 금기사항조항으로 들어가야 하는지에 대한 근거자료로 종합정리 되어 반영되어야 한다.
상기에 언급 된 바는 약물개발의 방법적 측면에서 TPP 의 활용이며, 약물의 가치 및 기술이전 측면에서의 TPP 는 이 목표치가 어떤 기준으로 설정 되는지 이다. 시장에 진입할 목표 약물의 프로파일은 당연 현재 치료 약물의 1) Unmet Medical Needs 2) 시장에서의 경쟁약물 3) 표준치료 지침 4) 진입국의 허가 규제 환경 등 주요 요소가 반영되어 설정되었을 때 구매욕구를 충족시키는 제품이 될 수 있다.
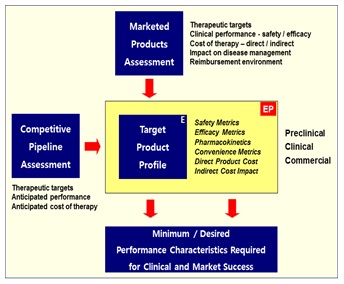
이렇듯 TPP는 약물개발 초기단계에 있어 Strategic Planning 의 기본이며, 개발 과정 중에는 개발의 방향에 대한 가이드를 제공할 뿐 아니라 약물의 인/허가 과정에 있어서는 FDA, EMEA 등 허가 규제기관의 Reviewer 들과 개발제품의 임상시험 허가 및 제품 등록을 위하여 논의 시 공유되어 상호 효율적인 Q&A 를 위하여 규제기관과의 교신을 위하여 준비되면 좋은 자료이다.
하기 표를 보면 약물의 개발 과정 속에 규제기관과의 교신 시 최초 작성된 TPP 는 개발의 다듬단계 미팅 시 논의에 근거가 된어 Pre-NDA 단계에 이르게 되면 거의 제출자료 목록의 전반을 cover 하여 Label 을 support 하는 완전한 단계에 이르게 된다.
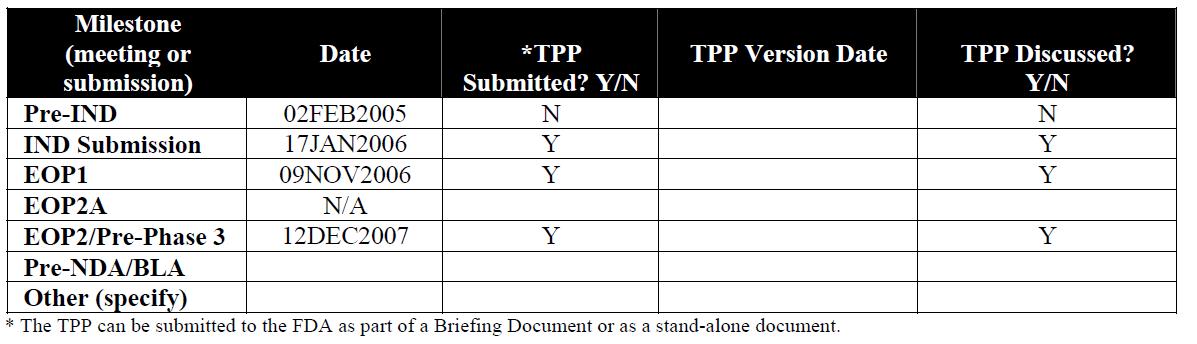
이 TPP 는 의무는 아니지만 거의 대부분의 제품허가를 위한 교신은 이 TPP 를 바탕으로 이루어진다. 허가 기관과의 첫 교신 시 작성되는 TPP 는 아주 primitive 하고 아직 채워야 할 항목이 많은 자료라면 개발이 진행되는 과정 속에 자연스럽게 허가를 support 하는 자료로 변모될 것이며 1시간 남짓의 짧은 Pre IND 미팅 시 개발사의 질의가 일목요연하게 정리되고 그 속에 분명한 규제기관의 의견이 반영될 수 있는 좋은 도구로 잘 활용된다면 성공적인 개발에 도움이 되지 않을까 한다. 등대가 없는 항구로 진입하는 배는 정확한 방향뿐 아니라 암초라는 risk를 만나 난파 될 수 있을 것이다. 그러나 TPP라는 등대의 불빛을 가이드 삼아 항구로 진입한다면 보다 안전하고 빠르게 항구에 정박할 수 있는 원리와 같다.
2. 허가자료의 작성은 글로벌 수준으로 개발의 초기단계부터 실시하여야 한다.
Initiate CTD documentation with global standard as early as possible in the development.
약물개발의 마지막 단계는 허가이고, 허가를 위하여 자료의 제출은 필수적이다.
그런데 허가자료 작성은 Common Technical Document 에 준하여 작성되어야 한다.
일반적으로 CTD 는 허가자료의 작성을 ICH 기준 하에 Harmonize 하여 진입하는 나라마다 허가자료를 다시 작성해야 하는 번그러움을 해소하고, 그 구성은 5개의 모듈로 나뉘어져 1번 모듈은 나라별 특이적인 행정적으로 요구되는 정보를 기입 하고 2번 모듈은 3. 4 5번 모듈에 작성될 제조 (Quality). 비 임상 (Non-clinical toxicology and pharmacology) 그리고 임상 (Clinical) 의 결과가 종합적인 요약으로 들어가야 한다 정도만 알고 있다.
그러나 CTD 작성에는 기본적인 작성의 원리가 있다. 하기 [표 4] 에 요약하였다.
모듈 3 Quality 에 있어서는 제조공정 및 안정성에 대한 자료 이외 개발의 각 단계에 사용된 배치별 동등성이 기 확립된 기준 하에 관리. 규명되어야 하며 모듈 4 의 비임상 단계에서는 동물의 독성시험과정에서 발견된 독성현상이 실질적으로 사람을 대상으로 하는 임상에서도 나타나는지, 나타난다면 원인은 무엇인지. 특정한 환자집단에서만 나타나는지. 그렇다면 그 환자집단에서만 나타나는 이유가 무엇인지 등에 대한 Safety 추적들이 이루어 져야 하며, 그러한 추적의 결과들이 이후 Label 의 Contraindication 및 Adverse Effects 뿐 아니라 허가 후에도 Risk Management Plan 등으로 반영. 제출되어야 한다. 이외에도 배치마다의 불순물관리 및 종간의 대사체와 관련된 Safety 관련이슈들도 설명되어야 하며 또한 이 비 임상에서의 연구결과가 어떻게 임상에 반영되는지 모듈 3과 모듈 5를 Cross Reference 하면서 제조물의 Quality 와 독성/약리 및 임상결과들이 연계되게 설명되게 작성되어야 하며, 만약 불 일치되는 결과의 도출 시는 그 이유가 적절히 셜명 되어야 한다. 임상의 경우는 특히 진입하고자 하는 국가의 Ethnic Difference 를 고려한 임상단계에서의 설계가 이루어져야 각 국가 진입 시 나라별 등록의 Delay 를 방지할 수 있다.
 3. Guideline 에 대한 이해 (Full Understanding)가 필요하다.
3. Guideline 에 대한 이해 (Full Understanding)가 필요하다.
CTD 작성의 기본원리는 ICH Guideline 에 준한다. 각 모듈마다 작성의 원리는 각각의 ICH Guideline 에 준하여 작성되어야 한다. 그렇다면 가이드라인을 이해하지 못하고 있다면 약 개발의
지침을 모르는 것이고 더불어 자료의 Generation 및 기록의 내용도 Guideline 의 지침에 벗어날 수 밖에 없다. 허가 단계에서 고객의 자료를 받아 CTD 를 작성 하다 보면 특히 제조 즉 CMC 부분에 대한 자료가 허가자료 작성에 필요하게 준비되고 구비되어 있지 못한 것을 많이 발견한다. CTD 작성에 있어 Module 3 가 NDA 실패의 큰 이유중의 하나이다. 규제기관이 보는 제품의 Quality 중 가장 중요한 point는 위에서도 설명 되었듯이 개발 과정 중에 생산되는 여러 배치간의 동등성이다. 이 동등성은 어떻게 규명되어야 하는지, 또한 일부의 차이가 있다면 어떻게 비 임상 및 임상을 진행하는 동안 그 차이로 인한 유효성 및 안전성에 영향을 주는지에 대한 연구가 시행되어야 하고 허가 시에 근거자료를 바탕으로 규명되어야 한다.
4. 임상시험 프로토콜의 Special Protocol Assessment 와 Enriched Clinical Study Design 이 필요하다.
허가를 위한 임상시험은 처음 설정한 TPP 를 임상적으로 규명하는 개발의 마지막 단계이다.
더불어 허가용 임상시험의 프로토콜 디자인은 약물의 특정 피험자에 대한 Benefit to Risk 를 증빙할 수 있도록 디자인 되어야 한다. 임상시험은 유효성만을 증빙하기 위한 시험이 아니다. 약효가 있는 물질은 독성이 있다. 더불어 Label 에 등록하기 위한 약물의 용량 및 약물 상호작용 및 타 요인에 의하여 혹시 모를 과다 노출에 대한 예상되는 부작용도 모니터링 되고 규명 되어야 한다.
또한 단기간의 치료가 목표가 아니고 장기간 복용해야 하거나 만성적으로 복용해야 한다면 Commorbidity Status 에서의 약물의 효과도 검증되어야 하며, 약물의 Responsiveness 에 영향을 줄수 있는 Intrinsic & Extrinsic Factors 도 규명 되어야 한다. 이렇듯 많은 요소들이 규명되어야 한다면 도대체 얼마나 많은 임상을 하여야 할까를 생각해 보면 하나의 임상시험 디자인에 보고자 하는 요소들이 전략적으로 잘 반영된 Enriched Clinical Study Design 이 R&D 비용을 절감하며 결과를 수집할 수 있는 최선의 방법일 것이다.
5. 경험 있는 프로젝트 매니저를 기용하여 신약개발이 주도 되어야 한다.
신약개발은 수많은 전문 Process 들이 서로 상관되어 진행되는 복잡한 과정이며, 중간 중간 끊임없는 Go/No Go Decision 이 이루어져야 하며 또한 Risk Management 도 이루어져야 한다. 우리는 세월호 사건을 보면서 Leader 의 선택이 많은 생명을 구할 수도 잃을 수도 있다는 사실에 대하여 다시 한번 통감했다. 그러나 우리나라 제약기업의 신약개발 구도를 보면 많은 과정들을 한 눈에 들여다 보고 Coordinate 해야 하는 프로젝트 매니저가 생산팀, 분석팀, 개발팀, RA 팀으로 병렬형으로 운영되는 구조 속에서 TPP 라는 Goal 을 향하여 Back and Forth 의 Decision Making 을 이루는 Coordination 은 이루어 질 수 없다. 또한 우리나라의 신약개발의 역사는 짧기 때문에 성공적인 개발을 이루어낸 프로젝트 매니저를 구하기란 또한 쉽지 않다는 것도 안다.
신약개발에 있어 프로젝트 매니저의 중요성은 이제는 널리 인식되어 프로젝트 매니저 양성 교육 및 제약개발 RA 실무자 양성 등 다양한 교육이 이루어지고 있으나 실질적으로 전체를 어우르는 지휘는 개개의 개발 업무 지식이 기반이 되어야 한다. 이런 업무의 기반은 또한 가이드라인의 이해에서 비롯된다.
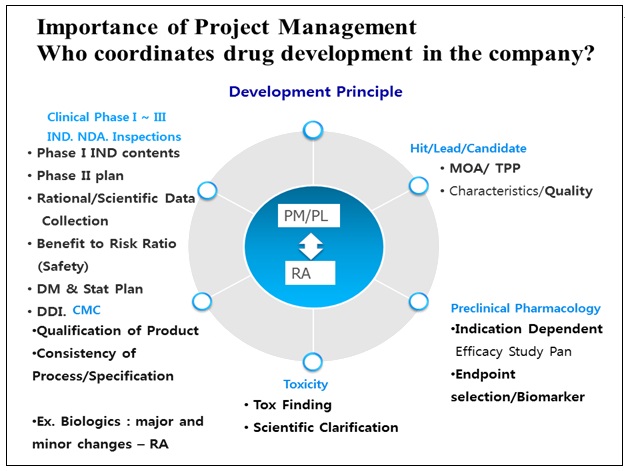
그간 신약개발의 프로젝트 매니지먼트 실무를 진행하면서, 기술이전을 중계하면서, 임상시험 허가 자료를 작성하면서, CRO 에서 근무 하면서 그리고 현재는 인/허가 전문 컨설팅 CRO를 운영하면서 이런 점이 반영된다면 좀 더 나은 제품개발이 이루어지지 않을까 하는 생각을 몇 가지 공유하였다.
요즘 많은 기업들이 임상 2상을 마치고 Value 를 올린 후에 3상 단계에 기술이전을 계획하며 개발을 진행하는 경우가 많다. 이러한 경우 개발이 TPP oriented 되게 개발되어 왔고 또한 구매자의 Unmet Needs 를 만족하는 제품이라면 또한 그 과정들이 Inter-related 되게 목적지향적으로 well documented 되고 TPP 하에 better presented 된다면 성공적인 기술이전이 가능하지 않을까 하며 마지막으로 성공적인 기술이전. 가치 있는 제품개발, 또한 성공적인 Global NDA 를 위하여 신약개발을 진행하는 분들에게 다음과 같은 Key Words 를 남기고 마무리 하고자 한다.
Define the goal (begin with the goal with unmet medical needs-public health value)
Develop a plan working backwards from the goal
Define level of benefit/risk
Determine interim steps that have to be achieved
Demonstrate clear target patient population
Efficiently and timely as possible
Strategic scientific development plan
Critical importance of predictive safety & efficacy biomarker which identify patient and to predict progress and response
Each component is part of whole strategy
Efficacy “threshold”
Tolerability profile
Foundation for basic preclinical-clinical consistency
Pathology of disease reasonable well understood
Animal models available
Animal models predictive of human disease
Do document with CTD format
Make your own TPP
Communicate with regulatory body
Special Prototocol Assessement
Strategic planning of statistics before clinical trial |
