사례 중심의 규제과학 현황과 나아갈 방향
‘CMC-Biologics; Minimal vs QbD Approach’
㈜바이넥스 연구개발본부장 이정진
1. CMC와 CTD
CMC란 Chemistry, Manufacturing and Control의 약어로 의약품 개발과정에서 의약품(원료, 완제)의 품질과 연관된 연구개발 및 제조공정이 일관성 있게 조절 및 관리되고 있음을 문서화하여 입증하는 것을 말한다.
합성 의약품이나 바이오 의약품의 품목 허가를 득하기 위해서는 규제기관에 품질 및 안정성, 비임상 및 임상 시험을 통한 안전성 및 유효성을 입증하기 위한 기술문서를 제출해야 하며, 국가간 공통으로 적용 가능한 국제 공통기술문서인 CTD(Common Technical Document) 양식에 따라 따른다. 그림 1의 CTD 삼각형에서 보여지는 바와 같이 CTD는 5개의 모듈로 이뤄져 있고, 이 가운데 CMC에 해당되는 부분은 모듈 3 품질(Quality)이다. CTD에서 CMC가 차지하는 비중 및 중요성은 비임상시험 및 임상시험 보고서 자료와 동등하다 할 수 있다.
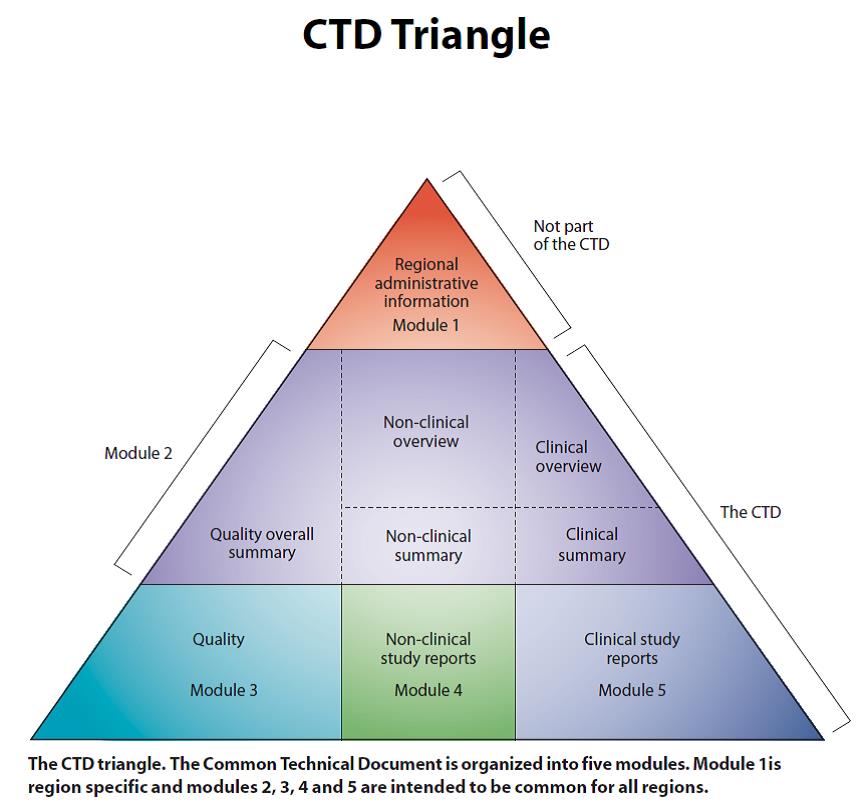
그림 1. CTD삼각형. 모듈 2~5 는 국제공통으로 사용 가능.
모듈 3 품질 부분은 다시 크게 원료의약품 (DS; Drug Substance or API; Active Pharmaceutical Ingredient)과 완제의약품 (DP; Drug Product)으로 나누고, 원료의약품의 경우, 일반정보, 제조(공정, 원료물질, 공정개발 및 검증 포함), 특성분석(구조 및 특성, 불순물 분석), 원료의약품관리(기준, 시험방법, 시험법 검증, 배치 분석, 기준설정 근거), 표준품관리, 용기(container closure system), 안정성 시험 자료를 포함한다. 완제의약품의 경우, 조성, 의약품개발(주성분과 부형제, 제형 개발, 과충전, 물리화학적 및 생물학적 성질 포함), 제조공정개발, 제조공정관리 및 공정검증, 주성분관리(기준, 분석법, 분석법검증, 기준설정근거, 배치분석 포함), 부형제관리(기준, 분석법, 분석법검증, 인체 또는 동물유래물질, 신규 부형제 포함), 표준품, 용기, 안정성 시험 자료를 포함한다.
2. QbD(Quality by Design)란?
QbD(품질설계) 용어는 Juran의 1992년 저서, JURAN ON QUALITY BY DESIGN(Subtitle: The New Steps for Planning Quality into Goods and Services)으로부터 대중들에게 알려지게 되었고, 제품의 출하시험에 의한 회고적인 품질관리(Quality by Test) 보다는 선제적으로 위험요소가 어디에 있는지 식별하고 이를 회피하기 위한 계획 하에 품질관리(Quality by Design)가 이뤄져야 한다는 개념이다.
의약품 제조에 있어 QbD 의 개념은 ICH(International Conference on Harmonization) 가이드라인 Q8(R2)에 정의되어 있는데, 과학적 지식과 품질 위해 관리(Quality Risk Management; QRM)를 기반으로 제품, 공정의 이해, 공정 관리에 중점을 두어 미리 설정된 목적을 가지고 시작하는 의약품 개발의 체계적 접근이다.
그러므로 의약품 개발에 있어 QbD 접근을 하면, 제품과 공정에 대해 보다 잘 이해할 수 있으므로 기업의 입장에선 제조 공정의 효율을 높이고, 비용 및 시간을 절약할 수 있다. 또한, 품목 허가 후 변경사항에 대해 보다 유연하고 효율적으로 대처할 수 있다. 한편, 규제기관의 경우 변경관리(Change Control) 횟수가 감소하여 그에 따른 자료검토 업무가 감소하며, 환자의 경우, 보다 일관된 품질의 안전하고 효과적인 제품 수혜가 가능하다.
3. Minimal 접근과 QbD 접근
의약품 개발에 있어 QbD에 의한 접근과 기존의 최소한(Minimal) 접근의 차이점을 비교하면, 표 1과 같이 요약할 수 있다.
먼저 전반적인 의약품 개발의 관점에서 기존의 최소 접근은 주로 실험적으로 한 번에 한 변수만을 연구개발한 반면, QbD 접근은 의약품의 주요품질속성(CQAs; Critical Quality Attributes)이 원료물질 속성(material attributes)과 공정 변수(Process Parameters)와 어떻게 연관되는지를 체계적으로 여러 가지 변수를 동시에 실험계획법(DoE; Design of Experiment)에 의해 통계적으로 접근하여 디자인 공간(Design Space)을 확립한다.
표 1. Minimal 접근과 QbD 접근 비교 (ICH Q8(R2)).
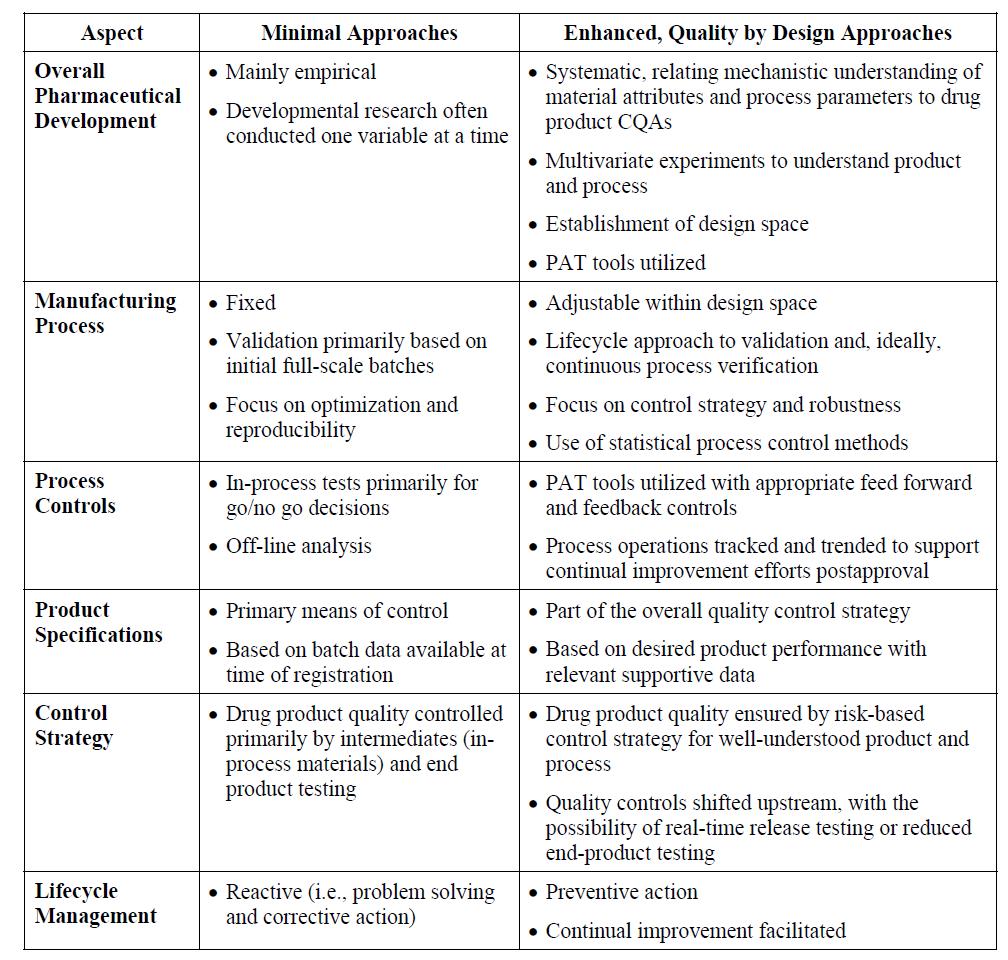
제조 공정 측면에서는 기존의 경우 공정개발 후 공정을 고정하고 초기 연속 3배치에 대해 검증하며, 재현성에 초점을 둔 반면, QbD 접근은 공정이 디자인 공간 내에서 조정 가능하며, 지속적인 확인을 통한 검증이 이뤄지며, 배치간 재현성보다 품질관리전략(Control Strategy)에 초점이 맞춰져 있다.
공정 관리 측면에서는 기존의 경우 Offline 분석을 통해 공정의 Go/No go 결정에 필요한 공정중시험(In-Process Testing)을 진행한 반면, QbD 접근은 Feed-forward 또는 Feed-back 관리를 위해 PAT(Process Analytical Technology) 도구가 사용되며, 허가 후에도 지속적인 품질 향상을 위해 공정을 추적하고 경향을 분석하게 된다.
기존의 경우 의약품의 규격이 품목허가자료에 제출된 배치 분석 결과에 기반한 주된 품질관리 수단인 반면, QbD 접근에서 규격은 관련된 지원 자료와 더불어, 원했던 제품 성과에 기반한 전반적인 품질관리전략의 일부이다. 완제 의약품의 품질관리도 기존에는 주로 중간체와 최종산물의 시험에 의해 이뤄진 반면, QbD에서는 보다 제품 및 공정에 대한 이해를 높이기 위해 위험기반 품질관리와 최종산물 시험에서 실시간출하시험(Real-Time Release Testing)이 가능토록 Upstream으로 이동되고 있다.
종합적으로 제품의 수명주기관리(Lifecycle Management) 측면에서 기존에는 문제 해결과 시정을 통한 반응적 조치인 반면, QbD 접근은 제품 수명주기 동안 지속적인 향상을 촉진하는 예방적인 조치이다.
4. QbD의 핵심요소와 과정
QbD의 실제적인 접근 과정 이해에 필요한 핵심요소는 QTPP(Quality Target Product Profile), CQA(Critical Quality Attribute), CPP(Critical Process Parameter), Design Space, PAT(Process Analytical Technology), Control Strategy, Real-Time Release Testing 을 들 수 있다(표 2).
표 2. QbD의 핵심요소 및 의미
| 핵심요소 |
의미 |
| QTPP(Quality Target Product Profile) |
의약품의 안전성과 유효성을 고려한 미래에 궁극적으로 도달코자 하는 제품의 품질 특성 |
| CQA(Critical Quality Attribute) |
원하는 제품의 품질을 보장할 수 있는 적절한 한계, 범위, 또는 분포를 가지는 물리화학적, 생물학적 또는 미생물학적 성질이나 특성 |
| CPP(Critical Process Parameter) |
CQA에 영향을 미쳐 원하는 품질의 생산공정을 보장하기 위해 감시 또는 관리해야 하는 공정 변수 |
| Design Space |
품질을 보증하기 위해 입증된 입력 변수(예: 원료물질속성) 와 공정 변수의 다차원적 상호작용과 조합. 디자인 공간 내에서의 작업은 변경으로 간주되지 않음. |
PAT
(Process Analytical Technology) |
최종 제품 품질을 보장하는 목표 내에서 CQA와 원료, 공정중, 또는 공정 속성을 적절한 측정을 통해 설계, 분석, 조절하기 위한 체계 |
| Control Strategy |
공정 성능 및 제품 품질을 확신할 수 있는, 제품과 공정 이해로부터 기인한 계획된 관리요소의 집합 (원료물질, 원료의약품 및 완제의약품과 연관된 속성 및 변수, 설비 및 장비 작동 조건, 공정중 관리, 최종제품 규격, 시험법, 감시 및 관리 주기 등) |
| Real-Time Release Testing |
공정 데이터(측정된 물질 속성 및 공정 관리의 검증된 조합)에 기반하여 공정중 또는 최종산물의 품질을 평가하고 보장할 수 있는 능력 |
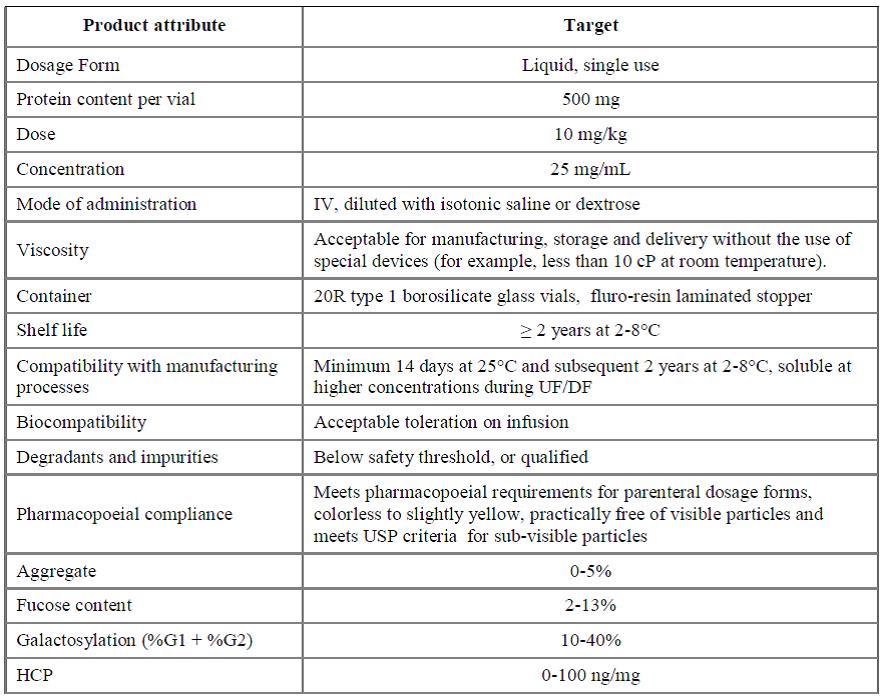
QbD 접근을 위해서는 무엇보다 개발하고자 하는 제품의 품질특성을 규정하는 QTPP의 설정이 우선되어야 한다. 요즘 가장 활발히 개발되고 있는 항체의약품의 QTPP 예시는 표 3과 같다. 다음은 품질에 영향을 많이 끼치는
표 3. QTPP for A-Mab
주요품질속성, 즉 CQAs를 결정해야 한다. 항체의약품의 경우, Aggregation, HCP(Host Cell Protein), 단당류 및 시알산 함량 등이 CQAs로 간주될 수 있다. CQAs가 결정되면, 원료물질속성과 공정 변수를 CQAs 와 연결하여 위해성 평가(Risk Assessment)를 수행한다. 이러한 위해성 평가는 각 단계별로 즉, 공정개발, 공정특성분석, 공정성능확인 과정에 필요에 따라 적절히 행해질 수 있고, 위험 정도를 수치화 [임계위험도(Risk Score; Criticality) = 영향력(Impact) × 불확실성(Uncertainty)]하여 평가한다. 그 결과를 종합하여 제품과 공정에 대한 디자인 공간을 개발하고 품질관리전략을 설계하고 시행한다. 궁극적으로 제품의 품질은 제품수명주기를 통해 지속적으로 향상시킨다(그림 2).
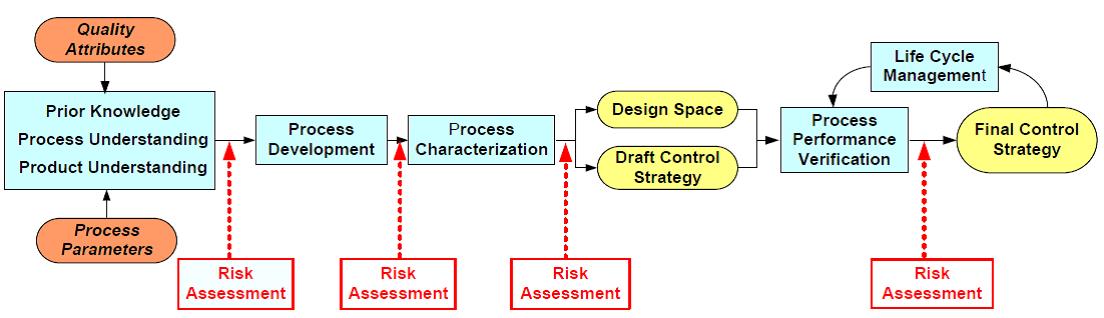
그림 2. QbD 패러다임을 통한 위해성 평가 단계
5. 결론 및 전망
본 기고를 통해 CMC가 CTD에서 차지하는 위치와 중요성을 언급하고, 공정과 제품 개발을 통해 CMC 자료를 준비하는 Minimal 접근과 QbD 접근에 대한 개념과 차이점을 언급하였다. QbD 접근의 많은 장점으로 말미암아 선진 규제기관(EMA & FDA)은 QbD 접근을 선호하고, 제약업계를 독려하고 있는 실정이다. 국내 바이오의약품 개발 업체들도 컨소시움 형태의 Working group이나 컨퍼런스, 세미나, 포럼 등을 통해 빠른 시간 내에 CMC의 일부분이라도 접목해 나가는 노력이 필요하리라 여겨진다.
ICH Q8(R2): Pharmaceutical Development (Nov 2009)
ICH Q9: Quality Risk Management (Nov 2005)
ICH Q10: Pharmaceutical Quality System (Jun 2008)
A-Mab: a Case Study in Bioprocess Development, v2.1 (Oct 2009)
