
- 재단소식
- 연구•산업 동향
효율적인 Hit to lead 방법을 통한 신약개발 전략
연세대학교 융합오믹스의생명과학과/생명공학과 한균희
옛말에 “될 성부른 나무는 떡잎부터 알아본다”는 말이 있다. 이 이야기를 신약개발에 적용하면, 혁신신약(나무)을 개발하기 위하여 우수한 분자작용점 혹은 선도물질 (떡잎)을 잘 골라야 할 것이다. 이를 위하여 기술혁신 (technology innovation), 다양성 기반 연구 (diversity base discovery)를 진행하여 온 것으로 생각된다. 예를 들면, High throughput screening을 통하여 screening 할 수 있는 물질의 수가 획기적으로 증가하자, 회사들은 새로운 골격의 물질을 확보하기 위하여 조합화학 기술에 투자를 하는 동시에, 전 세계의 화학자들에게 접근하여 합성화합물들을 저가에 구입하려는 시도를 하였다. 이는 다국적제약회사들의 small molecule library collection이 생각보다 다양하지 않다는 고민에서 출발한다. 이들 제약회사들은 chemical collection diversity를 획기적으로 개선할 것으로 예상하여 대대적으로 조합화학에 투자하였었다. 그렇지만 조합화학은 화합물의 갯수의 양적 증가라는 지대한 공헌에도 불구하고, 예기치 못한 문제점을 나타나게 된다. 예를 들면, 조합화학의 한 분야인 Parallel synthesis는 공정의 기계화를 통하여 물질을 합성하기 때문에, 실제로 응용할 수 있는 합성방법이 제한된다. 따라서 얻어지는 물질의 수에 비해서 diversity는 실제로 감소하는 경향이 있다. 그리고 조합화학에서는 reagent based synthesis를 하게 됨으로 인하여 Challenging한 구조의 화합물은 얻을 수 없는 단점이 생겼다. 그러므로 화합물의 수와 방법의 수월성만을 고려한 합성의 설계는, 만들기 쉬운 물질과 물성이 전혀 고려되지 않은 물질들을 양산하였다. 이로 인하여 실제로 약물의 개발이라는 측면에서는 초기비용을 더 지불해야 하는 상황을 초래한 것으로 생각된다.
실제로 새로운 혁신신약을 개발하기 위해서는 혁신적인 분자작용점과 신약으로 개발할 가능성이 높은 workable한 물질에서 시작해야 한다. 소위 “떡잎부터 다른” 과제를 고르기 위해서는 작용점 (molecular target)과 선도물질 (lead compound)에서 떡잎부터 다른 것을 골라야 한다. 그런데 혁신적인 분자작용점을 고를 수 있을 정도로 형편이 좋은 회사나 연구소는 한국에서 드물 것으로 예상된다. 이는 의생명 분야에 지속적인 연구와 관심을 통해서 선점해야 하는 성격으로 판단된다. 이 글에서는, 실제로 “고를 수 있는” 선도물질 (lead prioritization)에 대한 견해를 말씀 드리고자 한다.
“신약개발은 공정이다”라는 말이 있다. 신약개발은 작용점의 도출, 선도물질의 도출, 후보물질의 도출이라는 선택의 공정과, 선택된 “물건”에 대한 전임상 및 임상 평가를 통한 Go/No-go의 결정의 공정으로 나누어 진다. 이 가운데 선택할 수 있는 시기는 전임상연구 이전의 공정만이 가능하다. 실제로 한국적인 실정에서는 분자작용점은 선택의 문제가 아닌 경우가 많으므로, 선도물질과 후보물질의 선택 외에는 다른 선택은 없는 셈이다. 이 가운데 선도물질의 선택은 후보물질의 선택에 비해서 그 중요성이 낮게 인식되는 경향이 있다. 이는 최적화라는 공정을 거쳐서 좋은 후보물질을 얼마든지 도출할 수 있다는 과도한 자신감 혹은 미신 (myth) 때문으로 생각된다. 그러나 “떡잎”부터 다른 물질을 선도물질로 선택하는 것이 project의 성패를 좌우하는 중요한 성공요인이다. 실제로 혁신적인 분자작용점을 갖고도, 물질의 한계를 극복하지 못하여 신약개발을 실패한 예는 부지기수이다. 또한 공학적인 개념에서는 신약개발 공정은 시간의 공정으로 인식하게 된다. 이 시간의 개념에서 벗어나야만 효율적이고, 효과적인 선도물질 도출의 성과물을 낼 수 있을 것으로 사료된다.
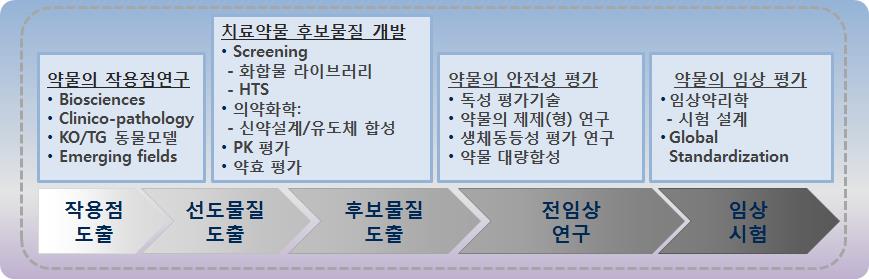
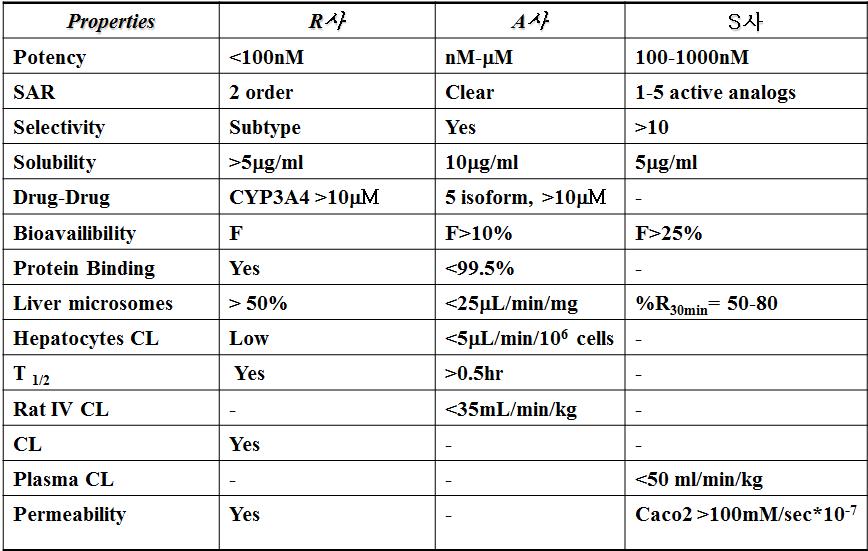
많은 다국적제약회사들은 screening으로부터 발견된 hit로부터 선도물질로 이행하기 위하여 다양한 criteria을 두고 있다. 예를 들면, 아래의 표와 같이 많은 다국적 제약회사들이 회사의 lead selection criteria 두고 우수한 선도물질을 도출하기 위한 Hit to Lead (H2L) process를 진행하고 있다. 본인이 근무하였던 회사 역시 비슷한 범주의 parameter을 위원회를 통하여 다양하게 적용하였다. 아래의 다양한 parameter는 공통적으로 Molecular property, Pharmacodynamics, Pharmacokinetics, structural optimization potential, patentability 등으로 구분할 수 있으며, 이 가운데 화합물에 직접적으로 연관된 parameter들은 molecular property, structural optimization potential 및 patentability 등을 들 수 있다. 다양한 library collection과 1,000,000개 이상의 화합물로부터 도출되고, 여기서 가장 좋은 것을 선택해서 신약을 개발하는 다국적 제약회사의 H2L 공정과는 달리, 우리의 현실은 라이브러리의 다양성도 수도 그리고 원천특허의 보호도 미약한 실정이다. 그러므로 단순히 선진국의 criteria 및 parameter를 그대로 원용하는 것보다는 우리 실정에 맞게 수정 및 변형 적용하는 것이 필요할 것으로 사료된다. 따라서 일반적으로 우리의 실정은 좋은 것을 선택하여 진행하는 방식이 아니고, 주어진 과제를 “반드시 성공시켜야 하는 실정”이므로, 실험실에서도 적용이 가능한 간단한 in silico 분석을 통해 가능한 몇 가지 제언을 하고자 한다.
화이자제약의 부사장을 역임하신 Lipinski박사의 “Rule of five”는 처음으로 화합물 분자의 구조적인 특성 (molecular descriptor)를 ADME property와 연결하여 설명한 것으로 많은 신약개발 연구자가 연구에 응용하고 있다.1 그러나 이 Lipinski’s rule은 아래와 같이 임상시험후보물질들의 molecular property가 ADME에 영향을 미치는 약물성 (druglikeness)을 평가한 것으로, 이 것을 직접적으로 선도물질 선택에 응용하기에는 무리가 있다. 그 뒤에 여기에 rotatable bond의 개수와 polar surface area (PSA)를 추가하여 좀 정밀하게 약물성을 점거하는 방법이 제시되었다.2
Lipinski’s Rule of Five: Parameters from clinical candidates
1) Mw ≤ 500
2) HDO (hydrogen bond donors) ≤ 5 (nitrogen or oxygen atoms with one or morehydrogen atoms)
3) HAC (hydrogen bond acceptors) ≤ 10(nitrogen or oxygen atoms)
4) An octanol-water partition coefficient LogP ≤ 5
5) An orally active drug has no more than one violation of the following criteria:
앞에서 언급한 약물성과 구분하여, 선도물질 선택에 응용하는 parameter들을 선도물질성 (leadlikeness)으로 부른다. 이 가운데 많이 인정받고 있는 있는 두 가지 rule를 소개하고자 한다. 첫 번째는 Amgen에서 사용되는 것으로 알려져 있는 것으로 아래와 같다.3 실제로 앞의 Lipinski’s rule에 비해서 분자량이나 cLogP값이 작은 범위에 있는 작은 물질을 선호하는 것을 알 수가 있다. 이는 선도물질 최적화하는 가운데 분자량이 커지면서 지용성 (lipophillicity)도 같이 증가하는 현실을 고려한 것으로 사료된다. 흥미로운 것은 단백질-분자상호작용에서 가장 큰 영향을 미치는 ionic interaction을 고려해서 pH7에서의 charged group을 고려한 것이다. 아울러 screening상에서의 방해자들을 제거하는 노력도 보인다.
– Mw = 200-350
– cLogP< 1.0-3.0
– Single charge present (secondary or tertiary amine preferred)
– Exclude chemically reactive functional groups:promiscuous inhibitors, frequent hitters, warheads
– Non-substrate peptides are suitable
두 번째는 GlaxoSmithKline (GSK)에서 발표한 것으로 자세한 parameter들은 아래와 같다.4 Lipinski rule에 비해서 좀 더 자세한 parameter설명이 되어 있으며, 흥미롭게도 LogSw scale을 도입해서 용해도의 중요성을 강조하고 있다. 이상의 rule에서 사용되는 molecular descriptor들은 ChemBioOffice 등 소프트웨어에서도 쉽게 계산이 가능한 것으로, 특별한 software package없이도 얼마든지 실험실에서 적용이 가능한 방법들이다.
– Mw ≤ 460
– -4 ≤ LogP ≤ 4.2, 0 ≤ LogD7.4 (LogP at pH 7.4) ≤ 3
– LogSw (the logarithm of the intrinsic aqueous solubility) ≥ -5
– RTB(Rotatable Bonds) ≤ 10, RNG (Number of Rings) ≤ 4
– HDO ≤ 5, HAC ≤ 9
Screening에서 발견된 hit들 가운데에서 선도물질로 선택하기 위해서는 구조변환을 통한 최적화의 용이성과 특허성을 우선적으로 고려하여야 한다. 원천특허가 확보된 회사 공유의 라이브러리에서 유래된 물질이 아닌 이상 (실제로 commercial source에서 유래된 라이브러리 세트, USA approved drug library 등이 대학이나 연구소에서 많이 사용됨), hit의 물질과 활성의 이력 (history)를 알아내는 것은 매우 중요하다. 기본적으로 화합물의 history는 SciFinder와 같은 화학검색엔진을 사용하면 되겠지만, 많은 정보로 인하여 혼란이 오는 경우도 있다. 이 경우 유용한 검색수단은 pubchem (http://pubchem.ncbi.nlm.nih.gov/)을 이용하는 것도 유용할 수 있다.
또한 small molecule이 상호작용하는 분자작용점을 안다면, molecular docking simulation이 가능할 것이다. 굳이 Accelrys software와 같은 고가의 프로그램을 이용하지 않더라도 AutoDock과 같은 Cambridge software의 ChemBioOfficepackage에 포함된 free ware를 사용할 수 있다. Docking simulation을 통해서 얻어지는 Binding mode를 분석하면 pharmacophore를 분석해서 virtual screening이 가능할 것이다. 특히 화학자적 관점에 hit의 chemical structure를 fragment로 분해해서 신물질을 설계하는 fragment based discovery도 가능할 것이다.5
참고문헌
1. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ.Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001,46 (1-3), 3–26.
2. Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45 (12), 2615–23.
3. Rishton, GB.Nonleadlikeness and leadlikeness in biochemical screening. Discovering. Drugs Today 2003, 8, 86-96.
4. Hann, MM,Oprea, TI.Current Opinion in Chemical Biology2004,8, 255–263.
5. Lipinski, C., and Hopkins, A. Navigating chemical space for biology and medicine.Nature 2004,432, 855-861.
이전
2013.12.02
다음
2014.02.03
