
- 재단소식
- 연구•산업 동향
안과질환의 유전자 치료제 개발 현황
(본 기고는 Nature Reviews of Drug Discovery, 2019년 6월호에 게재된 “Gene therapies in ophthalmic disease”의 내용을 바탕으로 작성하였다.)
유전성 망막 질환(Inherited retinal dystrophies, IRDs)은 유전자 결함에 따른 진행성 망막 변성의 희귀 질환을 모두 포함하고 있다. 환자군은 중증환자와 회생 불가능한 실명환자를 포함하며, IRD는 200개 이상의 유전자 결함과 관련이 있다고 알려져 있다.
스파크 테라퓨틱스사에서 개발한 럭스타나(성분명; Voretigene Neparvovec)는 FDA에서 승인된 첫 번째 안과질환 유전자 치료제로 이대립인자성(biallelic) RPE65 유전자 돌연변이가 원인인 IRD 성인 및 소아 환자를 대상으로 하고 있으며, 해당 치료제가 2018년 11월 EMA에서 승인된 이후 안과질환에 대한 유전자 치료제 개발이 본격화 되었다.
IRD는 유전자 치료제를 적용할 수 있는 매우 이상적인 질환이라고 할 수 있는데, IRD를 유발하는 원인 유전자를 명확하게 알고 있을 뿐 아니라, 안구가 Immune Privilege 영역이기 때문에 외부물질 유입에 따른 면역반응에서 자유로울 수 있기 때문이다.
현재 까지 진행된 임상시험 결과를 확인해 보아도 유전자를 전달하기 위해서 사용하는 아데노 부속 바이러스(Adenoassociated virus, AAV)와 렌티바이러스 벡터(Lentivirus vectors) 투여 후 부작용이나 면역 반응이 일어난 사례는 보고된 바가 없다.
IRD에서 가장 흔한 질환은 망막색소변성증(Retinitis pigmentosa), 맥락막결손(Choroideremia), 레베르시신경병증(Leber Hereditary Optic Neuropathy, LHON), 레버선천성흑암시(Leber’s Congenital Amaurosis, LCA), 스타르가르트 질환(Stargardt disease), 색맹(Achromatopsia)과 X-연관 망막층간분리증(X-linked retinoschisis, XLRS)이며 현재 개발되고 있는 대부분의 유전자 치료제는 해당 질환을 대상으로 하고 있다. 망막혈관질환(Retinal vascular diseases)과 노인 황반변성을 대상으로도 개발되고 있지만, 해당 질환의 경우, 단일 유전자의 결함으로 인해 발생되는 질환이 아니라 세포의 유전적 변이를 통해 생산된 단백질들이 복잡한 기전을 거쳐 발생하는 질환이다.
IRD를 대상으로 한 유전자 치료제는 보다 더 큰 안과질환 치료제 시장으로의 일보 전진으로 볼 수 있다. 럭스터나는 망막세포가 존재하는 환자를 대상으로 하고 있으며 투여경로는 망막하주사 (Subretinal injection)이다. 이러한 이유로 중증환자 대상에는 적용이 불가능하다.
안전성과 효능평가를 41명의 환자를 대상으로 진행하였고, 스파크 테라퓨틱스사에서는 시판 후 조사를 통해서 장기 안정성은 물론이고 이 외에도 잘 알려져 있는 부작용인 안구의 염증, 출혈, 통증을 지속적으로 추적 관찰 할 계획이다.
안과질환 치료제 시장분석
2017년 안과질환 치료제 시장의 총 매출액은 279억 USD로 전체 치료제 시장의 2.8%을 차지하였다. 안과질환의 치료제 시장은 지난 5년 동안 연평균 6.17%씩 지속적으로 성장하였지만, 대부분 신생혈관을 차단하는 치료제 위주의 시장이었다. 2017년도 한해 98억 USD의 매출액을 기록하였는데 이는 안과질환 치료에의 35%에 해당되는 수치이다.
신생혈관을 타겟하고 있는 치료제는 2013년에서 2017년, 지난 5년간 연평균 11.44%씩 성장하였고, 신생혈관을 타겟하고 있는 치료제 중 가장 많이 판매되고 있는 치료제는 루센티스( Ranibizumab, 제넨텍)와 아일리아(Aflibercept, 리제네론)로 2017년 한해에 각각 35억 USD와 62억 USD를 판매하였다.
루센티스는 2015년 이후 매출액이 지속적으로 감소되고 있는데, 이는 wet AMD 치료제인 아바스틴(Bevacizumab, 제넨텍)의 오프라벨 사용 및 혈관내피성장인자를 차단하는 제품(아일리아(Aflibercept, 바이엘), 아바스틴(Bevacizumab, 로슈)의 런칭으로 인한 영향으로 사료된다. (참고로 아바스틴은 AMD가 아닌 항암 치료제로 승인받았다.)
반면, 아일리아는 고공 성장세는 당분간 지속될 것으로 예상되는데 이는 당뇨병망막증(Diabetic retinopathy)을 포함하여 확장가능한 대부분의 적응증을 대상으로 승인을 받았으며, 루센티스 대비 투여횟수나 가격경쟁력 부분에서 우월성이 있기 때문이다.
하지만 루센티스의 특허가 2020년에 만료됨에 따라 바이오시밀러가 성공적으로 시장에 진입할 경우 혼란이 야기될 것으로 예상된다.
동공축소제(부교감 신경계 및 동공 팽창의 원인을 자극) 및 항녹내장 제제의 2017년 매출액은 60억 USD로 안과치료제 시장의 22% 차지하였으며, 안구건조 치료제는 45억 USD의 매출액을 기록하였다.
스파크 테라퓨틱스가 개발한 유전자치료제인 럭스터나는 출시된 2018년 첫해 6개월 동안 총 6.7백만 달러의 매출을 달성하였다. 스파크 테라퓨틱스가 미국에 해당 제품을 출시한 가격은 단안에 450,000 USD, 양안에 850,000 USD인데, 해당 가격은 Incremental cost- effectiveness ratio analysis에 의한 분석결과에서 제시한 가격 대비 50~75%보다 높게 책정된 금액으로 스파크사는 비용에 대한 부담을 줄이기 위하여 분할납부 프로그램을 운영하고 있다.
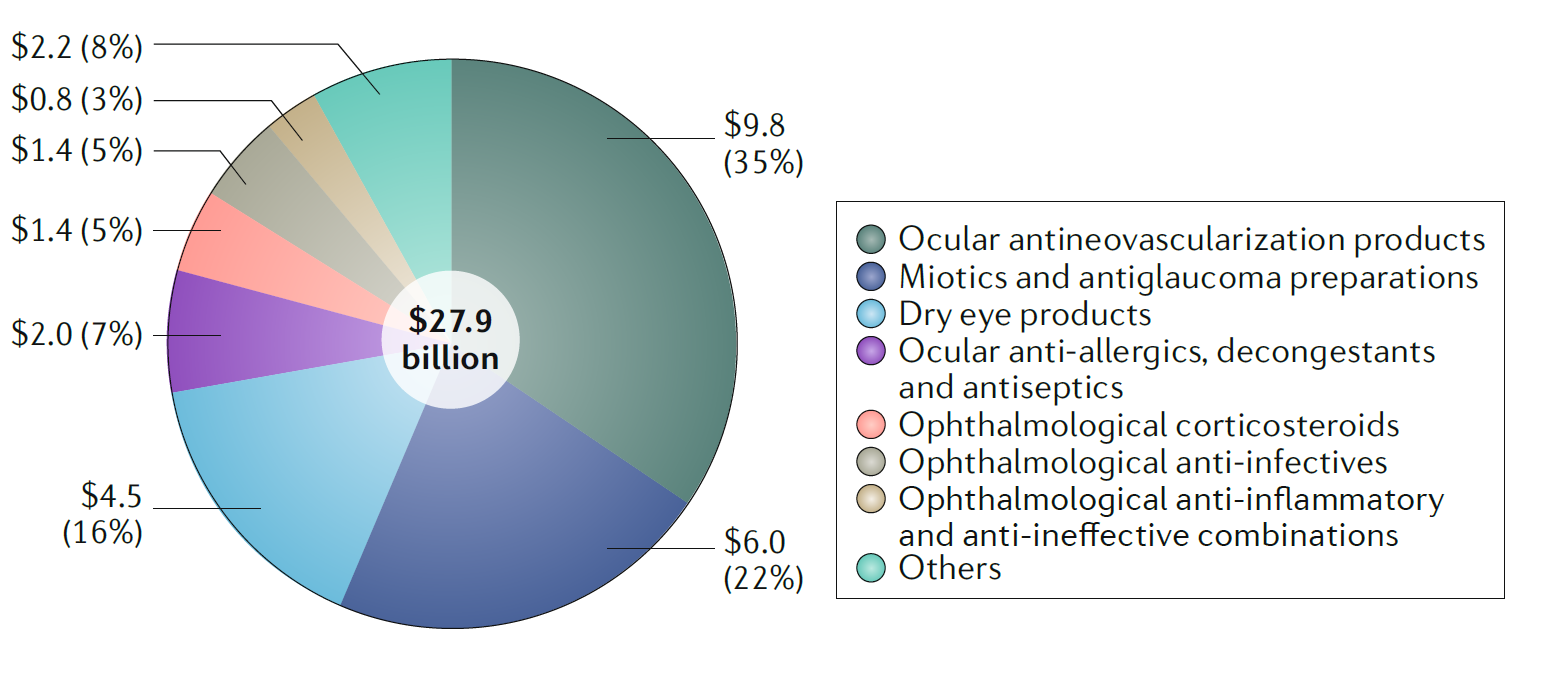
[그림1] 2017 안과질환 치료제 시장
파이프라인 분석
안구질환 유전자 치료제의 유전자 전달 방식은 타겟한 세포의 위치에 따라 달라진다. IRD의 대부분은 결함이 있는 유전자가 망막표피세포, 망막색소상피(Retinal Pigment Epithelium, RPE) 그리고 맥락막모세혈관층(Choriocapillaris)에 영향을 미치며, 이 경우 바이러스 전달체가 망막하 공간에 전달되어야 한다.
레버씨 시신경 위축증(Leber's Hereditary Optic Neuropathy, LHON)은 신경절 세포를 표적하고 있어서 망막층 내부로 잘 침투되기 위해서는 전달체(벡터)가 유리체강으로 전달이 되어야 하며, XLRS는 망막이 약하기 때문에 유리체 내로 전달하는 접근법이 선호된다.
현재 안과질환을 대상으로 한 유전자치료제의 개발 임상파이프라인(임상1,2,3상)은 총 25개 이다. 개발되는 임상 파이프라인(표 1)을 살펴보면, 막색소변성증 및 LCD와 같은 IRD를 포함할 뿐 아니라 AMD와 포도막 흑색종(Uveal melamoma)과 같이 안과질환 전체를 커버하고 있음을 확인할 수 있다.
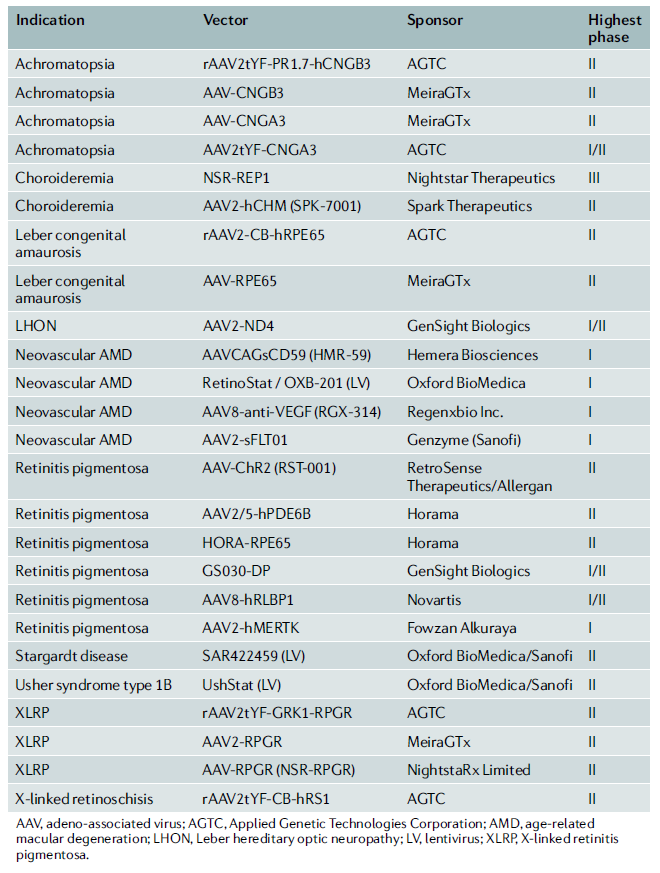
[표1] 안구질환 치료제의 임상파이프라인 현황
AGTC(Applied Genetic Technologies Corporation)와 MeiraGTx의 임상에 각각 5개 및 4개의 파이프라인을 보유하고 있어 안과질환 유전자 치료제를 선도주자로 자리 매김하고 있으며, 임상 2상에 진입한 파이프라인은 총 19개로 막색소변성증 5개, 색맹 4개, LDA 2개를 포함하고 있다.
대부분의 파이프라인은 변형된 유전자를 복구시키는 방향으로 개발되고 있으며, 주로 타겟하는 유전자는 REP1(encodes RAB escort protein 1), CNGA, CNGB, RPE65, RS1, RPGR 등이다.
가장 주목할 만한 파이프라인으로는 치료 단백질 유전자를 삽입시켜 신생혈관억제와 세포사멸을 억제하는 기전의 AMD 치료제와 레트로센스 테라퓨틱스사에서 개발하고 있는 망막색소상피 치료제 후보물질인 RST-001이다. RST-001는 channelrhodopsin 2를 전달함으로서 빛에 대한 민감도가 낮았던 세포에 광민감성을 부여하는 방식이다. 참고로 channelrhodopsin 2는 양이온을 신경세포 안으로 들어오도록 하여 신경세포의 활동전위 생성을 촉진하는 역할을 한다.
나이트스타(NightStar) 사의 NSR- REP1(AAV2-REP1)는 맥락막결손을 대상으로 한 치료제로 임상 3상을 진행하고 있는 유일한 치료제로써 가장 빠른 시일 내에 시판 승인을 기대해 볼 수 있다. 해당 후보물질은 눈 안에 REP1을 생성시키기 위해 재조합 인간 cDNA를 탑재한 AAV2 벡터이며, REP1은 세포 내 단백질 수송에 중요한 역할을 수행하는 단백질로 해당 단백질의 기능이 상실되면 비정상적인 세포내 단백질 수송이 이루어지면서 망막세포상피와 광수용체에서 노폐물을 제거하는 기전이 손상되어 맥락막결손이 진행된다. 해당 후보물질의 임상 3상의 연구는 2020년 1분기까지 140명의 글로벌 임상환자 모집완료를 목표로 하고 있다.
유전자치료제의 한계점
현재까지 승인된 유전자치료제(글리베라, 임리직, 럭스터나)들이 임상시험 성공이라는 신약개발에 있어 가장 큰 장벽을 넘었음에도 불구하고 여전히 생산과 임상 디자인, 장기 안전성 및 상업화 전략 등의 해결 문제가 남아있다.
유전자치료제의 경우, FDA에서는 IND 승인을 받기 전 CQA(Critical Quality Attributes)를 통제 할 수 있는 제조 프로세스는 물론 밸리데이션이된 분석방법을 제출하여야만 하는데 이는 유전자 치료제를 생산할 때 근본적으로 내재하고 있는 복잡성과 생산 사이클에 따라 나타날 수 있는 품질의 차이로 인하여 CQA를 완벽하게 통제하는 것이 요구되기 때문이다. 초기 전임상 연구 단계에서는 추가 biodistribution 연구 또한 요구되는데 이는 형질 도입한 유전자가 예상한 대로 발현되고, 혹시라도 비 표적 부위의 조직에 유전자가 전달되지 않았는지를 확인하기 위함이다. 초기 개발단계의 어려움을 극복하기 위해 FDA에서는 유전자치료제 개발자들에게 프리 IND 미팅이전에 INTERACT(Initial targeted engagement for regulatory advice on CBER products) 미팅을 통해서 해당기관과 논의 할 것을 권장하고 있다.
추가 임상연구에 대한 요구 및 임상연구개발의 시간지연을 막기 위해서는 세밀한 임상연구 개발 전략을 수립하는 것이 중요하다. 승인된 유전자약물은 임상I/II상 및 IIb의 결과에 따라서 FDA 승인이 촉진되기도 하므로 임상연구의 end point 및 환자 선정/제외 기준, 임상사이트의 선택이 매우 중요하다고 할 수 있다. 특히, 바이러스성 벡터 기반의 유전자 치료제는 안티바이럴-안티바디로 인해서 발생할 수 있는 문제로 인하여 적용 가능한 환자군이 매우 제약적일 수밖에 없다. 따라서 임상 디자인 설계 시 이에 대한 비용과 타임라인을 충분히 고려하여 설계하여야한다.
FDA와 보험사에서는 1차 end point 또는 supplementary evidence로 임상연구 기간 동안 추적 가능한 환자의 데이터를 요구 하고 있는 경우가 증가하고 있는 추세이다. 따라서 장기 안전성 확인과 함께, 진행하고 있는 임상환자의 추적데이터를 어떻게 확보할지에 대한 계획을 세우는 것이 해당 약물의 상업화 전략에 있어 매우 중요한 요소로 작용한다.<끝>
<참고문헌>
Gene therapies in ophthalmic disease, Nature Reviews of Drug Discovery, 2019.06
이전
2018.11.29
다음
2019.07.30